

 By Joy N. Ismail, PhD
By Joy N. Ismail, PhD
Rare disease exposes the limits of current clinical development models. Small, fragmented populations make patient identification difficult. Heterogeneous biology complicates endpoint selection. Limited precedent creates uncertainty in regulatory pathways. At the same time, the therapies being developed in this space are among the most advanced in medicine, often reaching patients before systems are fully equipped to support them.
These conditions have required the field to adapt. Trial design, patient identification, data infrastructure, and engagement models have all evolved under pressure. As precision medicine expands into more common diseases with genetically defined subpopulations, the same constraints are becoming more widely relevant. Rare disease surfaces these challenges earlier, but they are no longer confined to it.
One consequence is becoming clear. Trial success is largely determined before recruitment begins, through decisions made in protocol design, eligibility definition, and endpoint selection.
Speed comes from precision not volume
Traditional clinical development treats patient identification as a volume problem. The assumption is that broader outreach, more sites, and more screening will increase the likelihood of enrollment.
In rare disease, this approach introduces friction rather than efficiency.
Trials that randomize efficiently invest early in eligibility precision. Broad outreach paired with imprecise screening creates volume but not velocity. Patients who appear eligible in early stages but are later excluded increase burden on sites and on patients themselves, while slowing enrollment and introducing variability in the dataset.
This dynamic is amplified in rare disease. Small populations mean that every misclassified patient carries a higher cost, both operationally and statistically.
High-performing programs translate protocol criteria into clear, patient-ready screening logic before site activation. This includes structuring eligibility around data that can be validated upstream, such as genetic variants, clinical history, and digital prescreening inputs.
The impact of this approach is visible in practice. In programs supported by Sano that target rare genetic subtypes, structured education and prescreening contributed to high conversion to genetic testing, with the majority of screened patients progressing to eligibility confirmation. In parallel, patients who do not meet immediate criteria were retained within structured engagement programs, creating a reusable pool for future studies. These patients are not lost to the trial ecosystem. They become part of a persistent dataset that reduces future identification timelines and improves the efficiency of subsequent studies.
Eligibility precision functions as a control mechanism. It determines not only how quickly patients are identified, but whether the trial produces a usable signal.
Patient identification must start before the site
The difficulty of identifying eligible patients is often framed as a timeline issue. In rare disease, it determines whether a trial progresses at all.
Across clinical development, recruitment delays remain a persistent cause of inefficiency and failure. In rare disease, the issue is not only delay. It is the inability to reliably locate patients defined by specific genetic variants within fragmented healthcare systems.
Traditional site-based recruitment models are not designed for this. Expanding the number of sites increases cost and operational burden without improving precision.
Precision patient finding shifts the starting point from sites to data. Genomic datasets, electronic health records, and registries are used to identify likely eligible patients before site engagement.
The effect of this shift can be measured. In a program supported by Sano that targeted variants present in less than 3% of the population, combining pre-genotyped datasets with digital prescreening reduced identification timelines from six to twelve months to a matter of weeks, while lowering cost substantially.
In other programs, structuring eligibility criteria upstream reduces the number of patients who progress to site-level screening without meeting core requirements. This improves conversion rates to enrollment and reduces unnecessary burden on both patients and sites
The implication is that patient identification is no longer a recruitment function. It is a data and infrastructure function that determines both speed and feasibility.
Endpoint strategy is shifting from measurement to representation
In a recent episode of the Genetics Podcast, Derek Ansel, Global Vice President and Therapeutic Strategy Lead for Rare Disease and Oncology at Worldwide Clinical Trials, described endpoint selection as “the bane of rare disease.” His point was not only that endpoints are difficult to define. It was that endpoint choice, patient population, and operational feasibility are tightly coupled from the outset. A poorly chosen endpoint cannot be rescued operationally, and a broadly defined population can obscure signal to the point where a valid therapy appears ineffective.
This interdependence becomes more pronounced in heterogeneous conditions. Patients with the same diagnosis may differ significantly in symptom progression, baseline function, and response to treatment. A single endpoint is often insufficient to capture meaningful change across that variation.
Similarly, in a Sano-hosted Rare Disease Day 2026 webinar, clinical development experts and patient advocates emphasized that endpoint misalignment is not only a scientific issue but a practical one. Measures that are clinically convenient may fail to reflect what patients and caregivers experience as meaningful improvement. In some conditions, changes in sleep, behavior, or daily functioning carry more weight than traditional clinical markers, yet these are not consistently prioritized in trial design.
This creates a structural risk. Endpoint design is another pre-recruitment decision that determines not just what is measured, but whether the study can produce interpretable evidence. Trials can succeed on paper while failing to demonstrate real-world benefit, or fail statistically while improving outcomes that matter to patients.
As a result, endpoint strategy is shifting in two directions. First, there is increased use of composite and adaptive endpoints to better reflect heterogeneous populations. Second, there is earlier engagement with patient communities to define what constitutes meaningful change.
This shift also has regulatory implications. Accepting endpoints that better represent disease experience requires flexibility from regulators, particularly in conditions where precedent is limited.
Engagement is moving upstream into protocol design
Patient engagement is often discussed in the context of recruitment and retention. In rare disease, its primary impact is on protocol design.
Evidence shows that early patient involvement improves enrollment and reduces dropout. This is due to better alignment between trial design and patient constraints and expectations.
In the Rare Disease Day webinar, one panelist noted that many recruitment challenges are effectively determined at protocol lock. If visit schedules, procedures, and endpoints are not aligned with patient constraints, no amount of downstream effort will resolve the issue.
From an investment standpoint, highlighting the impact of patient-centric approaches can help secure buy-in and larger budgets, which are essential for scaling and ultimately adapting research to better serve both patients and scientific development. For example, a study revealed that investing in patient engagement can accelerate a pre-phase 2 product launch by 2.5 years, or a pre-phase 3 by 1.5 years.
The impact of patient engagement also extends beyond trial execution. Patient-informed design strengthens the evidentiary package by demonstrating clear medical need and appropriateness, supporting both regulatory confidence and downstream market access decisions.
Precision medicine is pushing regulation beyond indication-based models
Regulatory frameworks were built around discrete indications. Precision medicine, particularly in rare disease, is increasingly organized around mechanisms.
The FDA recently announced the Plausible Mechanism pathway for rare disease drug development. The idea is that a therapy targeting a specific biological pathway may be relevant across multiple conditions without requiring entirely separate development programs for each.
This represents a shift from indication-based to mechanism-based thinking. It has several implications.
First, it changes how evidence is generated. Data from one condition may inform development in another if the underlying mechanism is shared. This increases the value of translational and cross-indication datasets.
Second, it introduces new challenges for regulators. Evaluating therapies across multiple small populations requires flexibility in how evidence is interpreted, particularly when traditional randomized controlled trial designs are not feasible.
Third, it creates divergence across regions. While the US and UK have begun to formalize these approaches, equivalent frameworks are less developed in Europe. This can create friction and uncertainty for global programs.
The Rare Disease Day panel discussion emphasized that this lack of alignment has practical consequences. Differences in regulatory expectations affect where trials are run, how data is generated, and when therapies become accessible in different regions.
Regulatory systems are moving toward greater flexibility, but the pace and direction of that movement are uneven. For developers, this creates both opportunity and complexity. For patients, it creates variability in access that is currently built into the system.
Data infrastructure is becoming the bottleneck to scale
Precision medicine depends on the ability to identify, stratify, and follow patients across datasets. In rare disease, the limitations of current infrastructure are particularly visible.
Individual datasets are often too small to support robust analysis. Progress depends on linking data across institutions and geographies. However, data is frequently stored in incompatible formats, limiting interoperability.
The cost of this fragmentation is significant, both in financial terms and in lost opportunities for insight.
At the same time, the inputs are improving. Whole genome sequencing costs have declined dramatically, making comprehensive datasets more accessible. Standards such as FHIR and frameworks such as FAIR provide a foundation for integration.
The constraint is no longer data generation. It is data usability.
The implication is that infrastructure, not science, is becoming the primary limiting factor in scaling precision medicine. Sponsors that can integrate, standardize, and reuse data will be able to move faster and design more efficient trials.
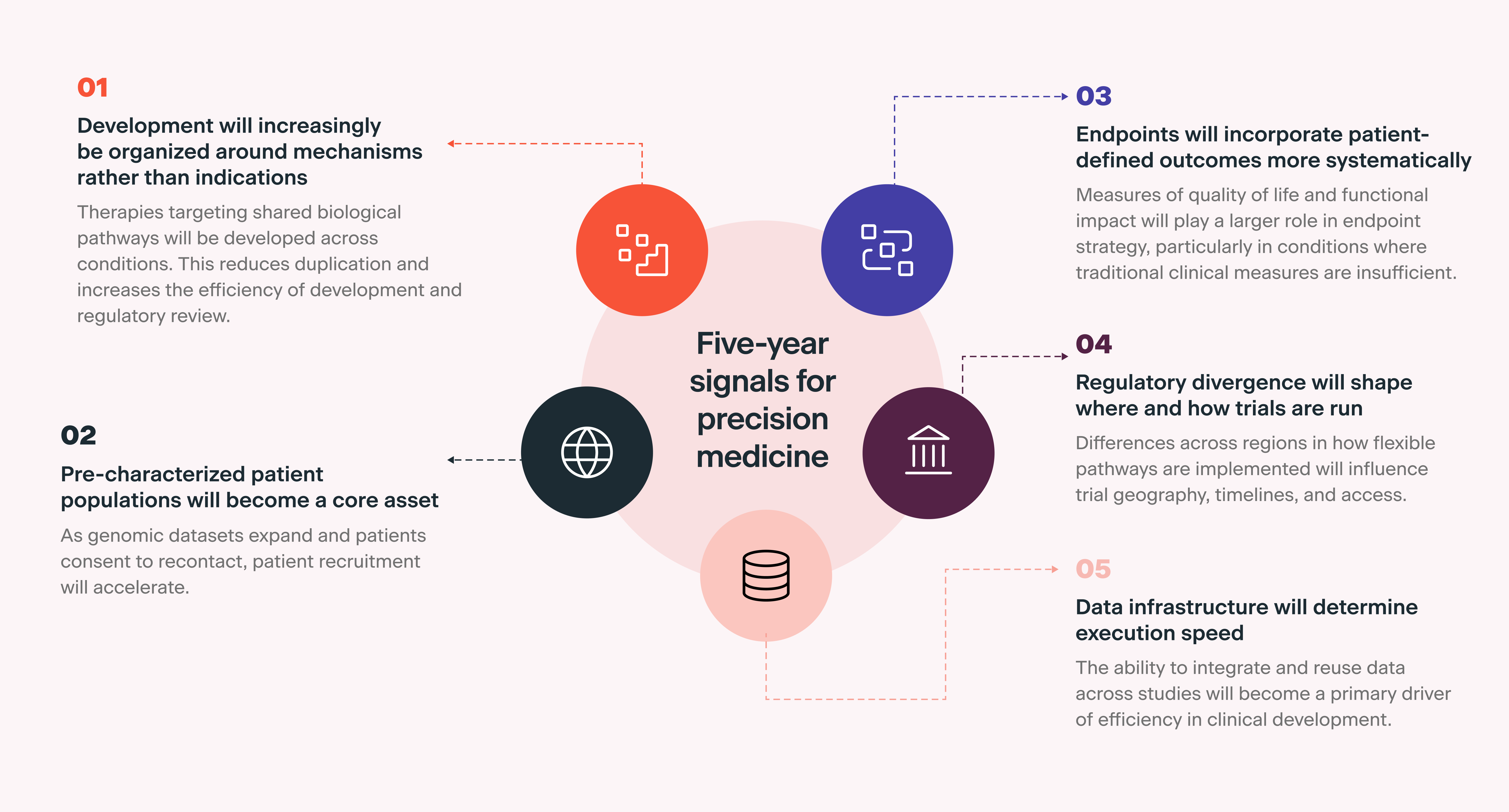
Five-year signals for precision medicine
Several patterns emerging from rare disease provide insight into how precision medicine is likely to evolve over the next five years. These are summarized in the infographic below.
Conclusion
Rare disease concentrates the challenges that precision medicine is beginning to encounter more broadly. The responses developed in this context provide a view of how clinical development is evolving.
Sponsors that treat rare disease as a model rather than a niche will be better positioned to design trials that generate credible evidence, secure regulatory confidence, and translate scientific innovation into real-world impact.
As these approaches scale, the defining difference between programs will be how early design decisions are made and how effectively they are integrated into the trial from the outset.